
ANTAGONISTAS DE LA VITAMINA K
Los dos anticoagulantes orales disponibles en España son acenocumarol y warfarina. Pertenecen al grupo de las cumarinas, no existiendo ningún representante del menos deseable grupo de las indanodionas.
Son dos medicamentos parecidos en sus características farmacocinéticas, desarrollando su acción máxima en 1-2 días, manteniéndose los efectos anticoagulantes tras la suspensión del tratamiento dos días para el acenocumarol y de dos a cinco días para la warfarina.
Los anticoagulantes orales se usan en tratamientos largos. En caso de precisar acción rápida comenzar con heparina al mismo tiempo que el derivado cumarínico. Retirar después progresivamente la heparina.
| RECUERDE
Los anticoagulantes orales son los medicamentos más citados en las obras especializadas en interacciones medicamentosas. Es conveniente estar familiarizado con las interacciones más importantes. Como norma general, tenga precaución en la administración de analgésicos y antiinflamatorios a pacientes bajo tratamiento anticoagulantes, porque las posibilidades de interacción son múltiples: desplazamiento de proteínas plasmáticas, efecto antiagregante plaquetario aditivo al anticoagulante, o efecto ulcerógeno que puede ser agravado por la hipocoagulabilidad sanguínea. |
HEPARINA Y DERIVADOS
Las heparinas son polisacáridos sulfatados que se obtienen de pulmón de bovino o de mucosa intestinal de cerdo. Son mezclas de cadenas de diferentes longitudes y la potencia anticoagulante depende del origen animal. En la práctica estas consideraciones no son importantes porque los preparados comerciales vienen estandarizados en Unidades Internacionales.
MECANISMO DE ACCIÓN
La acción anticoagulante se basa en la activación de la antitrombina III (AT III). La heparina se une a la AT III (para lo que necesita una secuencia específica de 5 monosacáridos) y produce un cambio estructural que aumenta la capacidad de la AT III para inactivar factores de coagulación, especialmente la trombina (factor IIa) y el factor Xa.
El mecanismo descrito basta para la inactivación del factor Xa. Para inactivar la trombina es preciso además la formación de un complejo triple trombina-antitrombina-heparina, en donde la heparina es -además de activador- el elemento de unión. Para ello es necesario que la cadena de heparina tenga un mínimo de 18 monosacáridos, de los cuales cinco deben ser la secuencia específica de activación mencionada más arriba. Prácticamente la totalidad de las cadenas de heparina convencional tienen más de 18 elementos (aunque sólo la mitad contiene la secuencia específica de activación) pero sólo del 25% al 50% de las cadenas de las heparinas de bajo peso molecular alcanzan esa longitud.
HEPARINAS CONVENCIONALES Y DE BAJO PESO MOLECULAR
Las heparinas se han utilizado tradicionalmente en la forma en que se extraen del tejido animal. Las más modernas Heparinas de Bajo Peso Molecular (en adelante HBPM) se producen fragmentando las cadenas de polisacáridos de la heparina convencional por distintos procedimientos (enzimático, químico o físico-químico según el fabricante). Las diferencias entre los dos tipos de heparinas se detallan en la tabla I.
La reducción de la longitud de las cadenas supone un cambio en el mecanismo anticoagulante. Por razones explicadas antes, las heparinas convencionales inactivan por igual la trombina y el factor Xa, pero las HBPM influyen preferentemente sobre el factor Xa. Aunque esta fue una de las principales razones para introducir heparinas de bajo peso molecular, la experiencia está demostrando que hay pocas diferencias en cuanto a eficacia o en incidencia de episodios hemorrágicos, y que son más interesantes las ventajas farmacocinéticas: la acción de las HBPM es más larga y con menos variaciones individuales, la biodisponibilidad es considerablemente mejor. Esto permite administrar 1-2 veces al día sin necesidad de ajustar la dosis por los parámetros de coagulación.
DIFERENCIAS ENTRE PREPARADOS
Hay en el mercado dos sales diferentes de heparinas convencionales, la heparina sódica y la heparina cálcica. Los efectos terapéuticos son equivalentes. Hay que tener en cuenta que la mayoría de las especialidades de heparina cálcica vienen acondicionadas para administración exclusivamente subcutánea[1]. La principal razón es que por esta vía es menos dolorosa que la sódica, aunque no hay inconvenientes en administrar heparina cálcica por vía IV. Sin embargo, deben elegirse para uso intravenoso heparinas donde se indique expresamente en el envase la idoneidad para dicha vía.
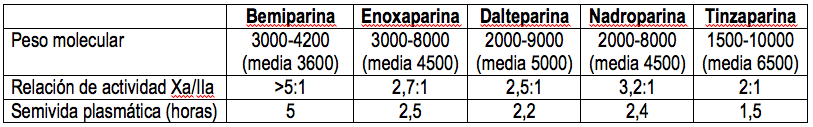
Las HBPM se diferencian en el origen de la materia prima y en el método de fraccionamiento. Sus características difieren algo, como se detalla en la Tabla II. A todos los efectos prácticos deben considerarse terapéuticamente equivalentes, pero no intercambiables.
[1] La administración de heparinas se hace por vía SC profunda o por vía IV (intermitente o preferiblemente en infusión continua). No usar la vía intramuscular.
TABLA I. DIFERENCIAS ENTRE HEPARINAS CONVENCIONALES Y HEPARINAS DE BAJO PESO MOLECULAR (HBPM)
|
Heparina |
HBPM |
|
| Peso molecular | 7.000-40.000 (media 12-15.000) | 3500-10000 |
| Núm. de monosacáridos | 23-130 | 10-20 |
| Relación de actividad Xa/IIa | 1:1 | 4:1-2:1 |
| Biodisponibilidad vía SC | 10-30% | 85-95% |
| Semivida vía SC | 3-18 h | 1,5-5 h |
| Forma usual de dosificar | Ajustando según el tiempo parcial de tromboplastina activada | Dosis fijas |
TABLA II. CARACTERÍSTICAS DE LAS HEPARINAS DE BAJO PESO MOLECULAR
USOS DE LAS HEPARINAS
Las heparinas se utilizan cuando se precisa acción anticoagulante rápida y de corta duración. En caso de requerirse hipocoagulación a largo plazo, se administra junto con anticoagulantes orales y se suspende la heparina (siguiendo con el preparado oral) cuando se estabiliza el tiempo de protrombina al valor deseado.
Se usan regímenes de dosis bajas de heparina subcutánea (usualmente 5000 UI cada 12 h) para la prevención de trombosis venosas profundas en enfermos hospitalizados de alto riesgo. En esta aplicación las HBPM (2500 U anti-Xa una vez al día, otras veces 50 U/kg una vez al día) son una alternativa a la heparina convencional, con eficacia y seguridad muy parecidas.
Están ampliándose las indicaciones de las HBPM a otros campos cubiertos por heparinas convencionales. Metanálisis de la experiencia clínica indican que en el tratamiento de trombosis venosas ya establecidas, las HBPM son como mínimo tan eficaces como la heparina no fraccionada, y tal vez sean mejores. De hecho, varias HBPM ya tienen autorizada tal indicación, y en el caso de fraxiparina, se acepta su uso en la profilaxis de las trombosis venosas en pacientes no quirúrgicos inmovilizados, cuya situación pueda definirse como de riesgo moderado o alto.
RECOMENDACIONES PARA LA ANTICOAGULACIÓN
| ENFERMEDAD | DURACIÓN DEL TRATAMIENTO | COMENTARIOS |
| Trombosis venosas profundas (TVP)
TVP proximal o embolia pulmonar |
– 3 meses para el primer episodio.
– 1 año para la primera recurrencia. – Crónico para sucesivas recurrencias. |
El régimen de baja intensidad con warfarina se adiciona al tratamiento inicial con heparina, la cual se suspende a los 2-3 días de que el INR esté en los valores prescritos. Si la warfarina está contraindicada, es efectivo un régimen de heparina SC cada 12 horas a dosis ajustadas para que el tiempo parcial de tromboplastina activada sea 1,5 veces el control. Los períodos recomendados de tratamiento se refieren a pacientes sin factores personales de riesgo. Si existen tales factores el tratamiento suele ser crónico. Las heparinas se utilizan cuando se precisa acción anticoagulante rápida y de corta duración. En caso de requerirse hipocoagulación a largo plazo, se administra junto con anticoagulantes orales y se suspende la heparina (siguiendo con el preparado oral) cuando se estabiliza el tiempo de protrombina al valor deseado. |
| Profilaxis de Trombosis venosas profundas (TVP) en pacientes de riesgo. | 8-11 días. | Se usan regímenes de dosis bajas de heparina subcutánea (usualmente 5000 UI cada 8-12 h) para la prevención de trombosis venosas profundas en enfermos hospitalizados de alto riesgo. En esta aplicación las heparinas de bajo peso molecular (HBPM), en dosis de 2500 U anti-Xa una vez al día y otras veces 50 U/kg una vez al día, son una alternativa a la heparina convencional, con eficacia y seguridad muy parecidas.
Sin embargo, en esta indicación, los derivados recombinantes de la hirudina, como desirudina (en dosis de 15 mg/12 h), parecen producir resultados profilácticos notablemente mejores que la heparina y las HBPM, reduciendo el riesgo de trombosis venosa profunda. |
| TVP de la pantorrilla, sintomática | Una semana de heparina IV seguida de tres meses de tratamiento de baja intensidad | El tratamiento anticoagulante preventivo sólo es necesario si no hay medios diagnósticos capaces de detectar la posible extensión de la trombosis. |
| Fibrilación auricular crónica (FA)
FA con miocardiopatía congestiva dilatada, o hipertrófica |
Crónico | |
| FA con cardiopatía valvular | Crónico | |
| FA con cardiopatía coronaria, hipertensiva o congénita | Evaluar individualmente el posible beneficio de la anticoagulación frente al aumento del riesgo de hemorragia. Los mejores candidatos son casos con insuficiencia congestiva (alto beneficio) y pacientes jóvenes (bajo riesgo).* | |
| FA por tirotoxicosis | Hasta estado eutiroideo y 2-4 semanas de ritmo sinusal. | |
| FA idiopática sin cardiopatía asociada | Crónico | Sólo en pacientes de más de 60 años con bajo riesgo de hemorragia. |
| Tras accidente vascular cerebral de origen cardioembólico | Tratamiento de alta intensidad por un año, después tratamiento de baja intensidad crónico. | El ácido acetilsalicílico puede ser una alternativa en caso de que los anticoagulantes estén contraindicados. |
| Cardioversión tras fibrilación auricular. | 3 semanas antes de la cardioversión (si es factible) y 2-4 semanas después. | La anticoagulación profiláctica está indicada si la fibrilación ha durado más de 3 días, en presencia de valvulopatía mitral o antecedentes de embolia sistémica. No suele ser necesaria en la cardioversión de otras arritmias auriculares. |
| Valvulopatías
Mitral o aórtica con fibrilación auricular crónica o paroxística. |
Baja intensidad.
Crónico |
Los casos no complicados, con ritmo normal, no suelen precisar anticoagulación preventiva. |
| Tras accidente vascular cerebral de origen cardioembólico. | Tratamiento de alta intensidad por un año, después tratamiento de baja intensidad crónico. | |
| Estenosis mitral con ritmo normal, aurícula izquierda hipertrófica (diámetro >55 mm). | Baja intensidad. Crónico. |
Algunos clínicos recomiendan también anticoagulación en caso de insuficiencia ventricular izquierda grave. |
| Prolapso mitral con ataques isquémicos transitorios. | La anticoagulación sólo está indicada si fracasa el control de los ataques isquémicos transitorios con ácido acetilsalicílico (500-1000 mg/día) | |
| Prótesis valvulares mecánicas | Régimen de alta intensidad. Crónico. | Usar régimen de baja intensidad de warfarina mas dipiridamol (5-6 mg/kg/día) si el régimen de alta intensidad está contraindicado. Añadir dipiridamol al régimen de alta intensidad si es insuficiente para controlar el embolismo sistémico. |
| Válvula mitral bioprotésica, con ritmo normal | Tres meses de baja intensidad tras el implante | Este tratamiento es opcional para válvulas aórticas bioprotésicas. Puede ser conveniente un tratamiento crónico con AAS. |
| Válvula mitral bioprostética, con fibrilación auricular o trombos en aurícula izquierda. | Tratamiento de alta intensidad por 3 meses tras el implante, seguido de tratamiento crónico de baja intensidad. | |
| Infarto agudo de miocardio
Anterior transmural, o inferior extenso con fibrilación auricular o insuficiencia congestiva. |
Tres meses tras el infarto. | El tratamiento anticoagulante va orientado a prevenir trombosis cerebrales o pulmonares. Para tratamientos preventivos del reinfarto o muerte repentina se suelen utilizar otros medicamentos, salvo que existan los factores especiales de riesgo que se citan después (ver tratamiento farmacológico del infarto de miocardio en grupo B06A1A). La utilización de anticoagulantes orales en la prevención primaria del infarto es objeto de controversia. Teóricamente, la inhibición de la cascada de coagulación y, por consiguiente, de la deposición de fibrina, puede proteger de un infarto de miocardio a los pacientes de alto riesgo. Sin embargo, los efectos adversos de los anticoagulantes orales son más importantes que los del ácido acetilsalicílico y la monitorización del tratamiento es bastante más compleja. Estos son motivos suficientes como para desaconsejar el uso de anticoagulantes orales en la prevención del infarto de miocardio. No obstante, en pacientes que no toleren o en los que esté contraindicado el ácido acetilsalicílico, puede ser una alternativa interesante. |
| Posinfarto con insuficiencia congestiva grave, fibrilación auricular o tromboembolismo venoso previo. | Crónico. | |
| Miocardiopatía idiopática dilatada. | Crónico. | |
| Prevención de trombosis cerebral recurrente. | Tratamiento de alta intensidad por un año, si no hay recurrencias pasar a tratamiento crónico de baja intensidad. | Sólo para casos recurrentes. Para prevención tras primer infarto o tras ataques isquémicos transitorios es preferible usar ácido acetilsalicílico. |
* En FA no reumática el ácido acetilsalicílico (325 mg/día) es eficaz como preventivo de trombosis. Es una alternativa que debe ser considerada en pacientes jóvenes sin factores adicionales de riesgo (el beneficio marginal de la anticoagulación es escaso), en casos de riesgo elevado de hemorragia cerebral (ej. mayores de 80 año), y cuando la anticoagulación no sea posible.
INTENSIDAD DE COAGULACIÓN
Baja intensidad: INR entre 2,0 y 3,0. Alta intensidad: INR entre 2,5 y 3,5.
INR = Ratio normalizado internacional. Los tratamientos de la tabla donde no se especifica la intensidad son todos de baja intensidad.
ANTIAGREGANTES PLAQUETARIOS (excl. heparina)
El proceso de agregación plaquetaria es una parte vital del complejo fenómeno de la coagulación. El proceso comienza con la adhesión de las plaquetas a la superficie subendotelial expuesta por la lesión vascular, y la posterior activación plaquetaria. La activación tiene como resultado final la formación de un entramado de plaquetas unidas entre sí por cadenas de fibrinógeno, que será el núcleo y estructura del tapón hemostático o del trombo.
La activación de las plaquetas comprende dos procesos principales:
- La formación y liberación de sustancias vasoactivas y participantes en el proceso de coagulación. Entre ellas destacaremos las que van a amplificar el proceso de agregación induciendo la activación de otras plaquetas: prostanoides (tromboxano A2), adenosina difosfato (ADP) y trombina.
- La aparición y activación de receptores de proteínas en la membrana plaquetaria. Los receptores son integrinas, una familia de moléculas que intervienen muy frecuentemente en interacciones célula-proteína. Entre los receptores plaquetarios se encuentra el responsable de la fijación de la plaqueta a la zona lesionada al reconocer y unirse a cadenas proteínicas del subendotelio (notablemente el colágeno y el factor de von Willebrand), pero a nuestros efectos nos interesa más el complejo de glucoproteína (GP) IIb/IIIa, que reconoce y fija las cadenas de fibrinógeno, formando la trama plaqueta-fibrinógeno-plaqueta del tapón hemostático. Al activarse la plaqueta aparecen en su superficie unos 50.000 receptores IIb/IIIa. Este receptor está estrechamente relacionado con el de ADP, de tal manera que el bloqueo del receptor de ADP impide la activación del complejo de glucoproteína IIb/IIIa.
El proceso de agregación es inhibido por varias sustancias naturales (generalmente prostaglandinas) que se producen en el endotelio y cuyo propósito es precisamente evitar que ocurra coagulación intravascular sin previa lesión.
Puede influirse de muy diversas formas sobre este complejo mecanismo. De hecho el grupo está constituido por medicamentos usados inicialmente para otras indicaciones y que han sobrevivido como antiagregantes por haber demostrado utilidad clínica, y por una serie de fármacos nuevos que se han concebido haciendo uso de los modernos conocimientos sobre el mecanismo de la agregación plaquetaria. Muchos de los mecanismos de acción de la clasificación siguiente se han descubierto con posterioridad al uso terapéutico.
– Actúan sobre los mediadores de la activación plaquetaria
- Inhiben la producción de tromboxanos: Ácido acetilsalicílico, triflusal
- Bloquean la acción de ADP: Dipiridamol, clopidogrel, ticlopidina.
– Bloqueantes del receptor GP IIb/IIIa:
- Anticuerpos monoclonales: Abciximab.
- Péptidos sintéticos: Eptifibátida..
- Estructuras no peptídicas: Tirofibán.
– Análogos de inhibidores naturales de la agregación: Epoprostenol (prostaciclina), iloprost.
Esta clasificación farmacológica no tiene gran utilidad para la selección de antiagregantes. La experiencia clínica es más importante. Los bloqueantes del receptor IIb/IIIa y los análogos de los antiagregantes naturales son mucho más potentes que los antiagregantes tradicionales, pero son productos inyectables cuya utilidad terapéutica está limitada por la vía de administración y por su propia potencia farmacológica. A medida que se profundiza en el mecanismo de la agregación plaquetaria, tanto más se borra la distinción tradicional (y un poco artificial) entre medicamentos anticoagulantes y antiagregantes plaquetarios.
Los antiagregantes se usan como preventivos de la formación de trombos en tres situaciones:
– Riesgo de episodios obstructivos coronarios y cerebrales.
– Cirugía vascular y diálisis
– Prevención de trombosis venosas profundas.
* En FA no reumática el ácido acetilsalicílico (325 mg/día) es eficaz como preventivo de trombosis. Es una alternativa que debe ser considerada en pacientes jóvenes sin factores adicionales de riesgo (el beneficio marginal de la anticoagulación es escaso), en casos de riesgo elevado de hemorragia cerebral (ej. mayores de 80 año), y cuando la anticoagulación no sea posible.
ANTIAGREGANTES PLAQUETARIOS (excl. heparina)
El proceso de agregación plaquetaria es una parte vital del complejo fenómeno de la coagulación. El proceso comienza con la adhesión de las plaquetas a la superficie subendotelial expuesta por la lesión vascular, y la posterior activación plaquetaria. La activación tiene como resultado final la formación de un entramado de plaquetas unidas entre sí por cadenas de fibrinógeno, que será el núcleo y estructura del tapón hemostático o del trombo.
La activación de las plaquetas comprende dos procesos principales:
- La formación y liberación de sustancias vasoactivas y participantes en el proceso de coagulación. Entre ellas destacaremos las que van a amplificar el proceso de agregación induciendo la activación de otras plaquetas: prostanoides (tromboxano A2), adenosina difosfato (ADP) y trombina.
- La aparición y activación de receptores de proteínas en la membrana plaquetaria. Los receptores son integrinas, una familia de moléculas que intervienen muy frecuentemente en interacciones célula-proteína. Entre los receptores plaquetarios se encuentra el responsable de la fijación de la plaqueta a la zona lesionada al reconocer y unirse a cadenas proteínicas del subendotelio (notablemente el colágeno y el factor de von Willebrand), pero a nuestros efectos nos interesa más el complejo de glucoproteína (GP) IIb/IIIa, que reconoce y fija las cadenas de fibrinógeno, formando la trama plaqueta-fibrinógeno-plaqueta del tapón hemostático. Al activarse la plaqueta aparecen en su superficie unos 50.000 receptores IIb/IIIa. Este receptor está estrechamente relacionado con el de ADP, de tal manera que el bloqueo del receptor de ADP impide la activación del complejo de glucoproteína IIb/IIIa.
El proceso de agregación es inhibido por varias sustancias naturales (generalmente prostaglandinas) que se producen en el endotelio y cuyo propósito es precisamente evitar que ocurra coagulación intravascular sin previa lesión.
Puede influirse de muy diversas formas sobre este complejo mecanismo. De hecho el grupo está constituido por medicamentos usados inicialmente para otras indicaciones y que han sobrevivido como antiagregantes por haber demostrado utilidad clínica, y por una serie de fármacos nuevos que se han concebido haciendo uso de los modernos conocimientos sobre el mecanismo de la agregación plaquetaria. Muchos de los mecanismos de acción de la clasificación siguiente se han descubierto con posterioridad al uso terapéutico.
– Actúan sobre los mediadores de la activación plaquetaria
- Inhiben la producción de tromboxanos: Ácido acetilsalicílico, triflusal
- Bloquean la acción de ADP: Dipiridamol, clopidogrel, ticlopidina.
– Bloqueantes del receptor GP IIb/IIIa:
- Anticuerpos monoclonales: Abciximab.
- Péptidos sintéticos: Eptifibátida..
- Estructuras no peptídicas: Tirofibán.
– Análogos de inhibidores naturales de la agregación: Epoprostenol (prostaciclina), iloprost.
Esta clasificación farmacológica no tiene gran utilidad para la selección de antiagregantes. La experiencia clínica es más importante. Los bloqueantes del receptor IIb/IIIa y los análogos de los antiagregantes naturales son mucho más potentes que los antiagregantes tradicionales, pero son productos inyectables cuya utilidad terapéutica está limitada por la vía de administración y por su propia potencia farmacológica. A medida que se profundiza en el mecanismo de la agregación plaquetaria, tanto más se borra la distinción tradicional (y un poco artificial) entre medicamentos anticoagulantes y antiagregantes plaquetarios.
Los antiagregantes se usan como preventivos de la formación de trombos en tres situaciones:
– Riesgo de episodios obstructivos coronarios y cerebrales.
– Cirugía vascular y diálisis
– Prevención de trombosis venosas profundas.
TABLA I. PRINCIPALES ANTIAGREGANTES PLAQUETARIOS
|
MEDICAMENTO MECANISMO DE ACCIÓN |
COMENTARIO |
Ácido acetilsalicílico, Triflusal.Inhibición irreversible (acetilación) del enzima ciclooxigenasa, que interviene en la síntesis de precursores comunes de tromboxanos (proagregantes) y prostaciclina, PGI2 (antiagregante). El predominio de la acción antiagregante se debe a que la prostaciclina es sintetizada por células endoletiales vasculares, capaces de producir nuevas moléculas de ciclooxigenasa tras la inactivación inicial por el ácido acetilsalicílico. Por el contrario, las plaquetas (que son fracciones celulares y, que por tanto, carecen de núcleo), son incapaces de producir nuevas moléculas de ciclooxigenasa, con lo que no se sintetizan precursores de los tromboxanos. |
El ácido acetilsalicílico (AAS) es el medicamento más experimentado. Se ha convertido en el antiagregante de elección y es el patrón de comparación del grupo. Las dosis como antiagregante están entre 75 y 325 mg/día. Dosis mayores no aumentan la eficacia pero sí los efectos adversos. Por su parte, el triflusal es un derivado trifluorado del AAS, con el mismo mecanismo de acción. Medicamento de investigación española. La documentación clínica es más limitada que con los anteriores, pero comienza a ser significativa |
DipiridamolEleva de niveles intraplaquetarios de AMP cíclico, impidiendo la acción de los mediadores de la activación plaquetaria. |
No se emplea como monoterapia porque el efecto antiagregante se obtiene a dosis que originan efectos secundarios frecuentes por la acción vasodilatadora. Se suele usar asociado al AAS o anticoagulantes en prevención de tromboembolismo en casos de contacto de la sangre con superficies distintas del endotelio vascular: prótesis vascular, «bypass» coronario o femoral-popliteal, prevención de trombosis venosas, etc. En la mayoría de las aplicaciones la asociación AAS+dipiridamol no ha mostrado ser más efectiva que el AAS solo. |
Ticlopidina, ClopidogrelBloquean la activación plaquetaria inducida por ADP, inhibiendo de forma selectiva la unión de éste a sus correspondientes receptores de la superficie plaquetaria. |
Son antiagregantes de eficacia similar o marginalmente superior al AAS pero con incidencia de efectos secundarios bastante mayor. Ticlopidina parece producir neutropenia como efecto secundario, con incidencia muy baja y usualmente en los primeros tres meses del tratamiento. Este riesgo parece ser algo menor con clopidogrel. Ambos pueden ser una alternativa interesante en pacientes que no toleren el AAS. Dosis de 75 mg/24 h de clopidogrel producen un efecto antiagregante plaquetario equivalente a 250 mg/12 h de ticlopidina. La primera parece tener un menor riesgo de inducir casos graves de neutropenia Se ha autorizado el uso de ticlopidina en prevención de oclusiones tras procedimientos coronarios quirúrgicos. Hay datos positivos sobre la eficacia de ésta en el manteniento a largo plazo de la funcionalidad de los injertos de vena safena en cirugía de derivación (bypass) de miembros inferiores, además de reducir la incidencia de episodios isquémicos no relacionados con el injerto. Esto resulta de especial interés dado que sin terapia coadyuvante, la tasa de funcionalidad llega a bajar a un 50% a los dos años para injertos femoropoplíteos o femorotibiales. |
Abciximab, Tirofibán, EptifibátidaAntagonistas de los receptores GP IIb/IIIa. |
Abciximab es un anticuerpo monoclonal que se acopla a los receptores GP IIb/IIIa bloqueándolos. Es un antiagregante muy potente que sólo puede administrarse por infusión IV. La incidencia de episodios hemorrágicos es alta. La indicación autorizada es la prevención de la obstrucción vascular tras angioplastia, en tratamiento aditivo al AAS y heparina. Tirofibán y eptafibátida también son administrados por vía IV y han sido autorizados para la prevención del infarto de miocardio precoz en pacientes con angina inestable o infarto de miocardio sin onda Q cuyo último episodio de dolor torácico se haya producido en las últimas 12 horas y que presenten cambios en el ECG y/o un aumento de los enzimas cardíacos. Se utilizan en combinación con heparina y ácido acetilsalicílico. |
IloprostSe trata de un análogo químicamente estable del epoprostenol (prostaciclina). |
Autorizada en España en la tromboangeítis obliterante avanzada (enfermedad de Buerger) con isquemia grave de las extremidades, en los casos en que no esté indicada la revascularización. También ha demostrado eficacia en la inhibición de la agregación plaquetaria inducida por heparina, incluso en casos refractarios al ácido acetilsalicílico. Asimismo, la administración IV de iloprost es eficaz en el tratamiento del fenómeno de Raynaud secundario a esclerodermia, produciendo una reducción de la frecuencia y severidad de los ataques. Además, previene e incluso llega a curar las úlceras digitales. |
Prevención de trombosis coronaria y cerebral
El concepto cubre las situaciones siguientes:
Alto riesgo.
– Infarto agudo de miocardio
– Posinfarto de miocardio
– Antecedentes de trombosis cerebral o ataques isquémicos transitorios (TC/AIT)
– Prevención en otros pacientes de alto riesgo (angina estable e inestable, diabetes, fibrilación auricular, claudicación intermitente, etc.).
Bajo riesgo.
– Prevención primaria en sujetos sin factores especiales de riesgo.
El ácido acetilsalicílico sigue siendo el agente más coste-efectivo para la prevención secundaria de la enfermedad isquémica cardíaca. Administrado en pacientes que han sufrido un infarto de miocardio, este fármaco reduce los índices de mortalidad y de infarto recurrente en un 20%. En pacientes con angina inestable, el ácido acetilsalicílico puede prevenir un 30% de los casos de infarto de miocardio. También los pacientes con injertos coronarios o sometidos a angioplastia, así como aquellos con angina estable, pueden beneficiarse de los efectos preventivos del ácido acetilsalicílico.
El uso de este medicamento para la prevención primaria de la enfermedad isquémica cardíaca ha sido sometido a debate durante mucho tiempo. Actualmente hay datos que demuestran que el ácido acetilsalicílico puede prevenir el infarto de miocardio en hombres.
En este sentido, se ha comprobado una reducción del 32% del riesgo de infarto de miocardio no mortal por año en sujetos de alto riesgo con ácido acetilsalicílico, frente a un 31% con hipolipemiantes de tipo estatina. Los correspondientes valores para pacientes de bajo riesgo son del 41% (ácido acetilsalicílico) y 37% (estatinas), todo ello en relación al placebo. Tras los correspondientes ajustes estadísticos, estos datos indican que el tratamiento puede prevenir un infarto de miocardio no mortal anual por cada:
|
Fármaco |
Pacientes de bajo riesgo |
Pacientes de alto riesgo |
| Acido acetilsalicílico | 643 | 347 |
| Estatinas | 311 | 263 |
Como se aprecia en la tabla anterior, en hombres sanos, el grado de protección que el ácido acetilsalicílico confiere frente a los infartos de miocardio no mortales es similar al de las estatinas. ¿Significa esto que debería prescribirse ácido acetilsalicílico a todo el mundo para prevenir el infarto de miocardio?
La pregunta debe ser contestada en negativo, ya que, para empezar los datos clínicos disponibles sólo se refieren a hombres (no a mujeres). Por otro lado, en las personas de bajo riesgo, el riesgo absoluto de infarto de miocardio es inferior a un 0,5% anual, por lo que cualquier medida preventiva (incluyendo la administración profiláctica de ácido acetilsalicílico) tendría un efecto muy marginal. Además, aunque este fármaco es muy bien tolerado en general, es capaz de producir, incluso con dosis pequeñas, molestias gástricas que, en casos raros, pueden convertirse en hemorragias digestivas o cerebrales. Aunque el riesgo de accidente cerebrovascular es muy bajo en términos absolutos, existe un exceso de riesgo de un caso por cada tres infartos de miocardio prevenidos, algo que no ocurre ciertamente con las estatinas.
Por todos estos motivos, sólo los hombres con alto riesgo de infarto de miocardio (fumadores, hipertensos, obesos, hipercolesterolémicos, etc) son candidatos a la prevención primaria con ácido acetilsalicílico, aunque sólo como una más de las medidas preventivas que es preciso adoptar en este tipo de personas (dieta, tabaquismo, actividad física, hipertensión, etc).
La administración de ácido acetilsalicílico (160-300 mg/día), iniciada dentro de las 48 horas del comienzo de los síntomas de accidente isquémico cerebral, ha demostrado reducir el riesgo de recurrencia de este tipo de accidentes cerebrovasculares y mejorar el pronóstico de este tipo de pacientes.
La utilización de anticoagulantes orales en la prevención primaria del infarto también ha sido objeto de controversia. Teóricamente, la inhibición de la cascada de coagulación y, por consiguiente, de la deposición de fibrina, puede proteger de un infarto de miocardio a los pacientes de alto riesgo. Sin embargo, los efectos adversos de los anticoagulantes orales son más importantes que los del ácido acetilsalicílico y la monitorización del tratamiento es bastante más compleja. Estos son motivos suficientes como para desaconsejar el uso de anticoagulantes orales en la prevención del infarto de miocardio. No obstante, en pacientes que no toleren o en los que esté contraindicado el ácido acetilsalicílico, puede ser una alternativa interesante.
Cirugía vascular y diálisis
En la prevención de oclusión tras cirugía vascular, el beneficio es del orden de 90 casos evitados por cada 1000 tratamientos (de siete meses en intervenciones coronarias y 19 meses en las periféricas). En pacientes bajo hemodiálisis pueden evitarse 220 oclusiones del acceso por 1000 tratamientos de dos meses.
Los riesgos son de un aumento de 13/1000 tratamientos de los episodios hemorrágicos importantes y de un posible aumento (no significativo estadísticamente) de 1 caso de hemorragia fatal por 1000 tratamientos. No hay diferencia significativa de eficacia si se comienza el tratamiento antes de la operación o inmediatamente después. Puede ser prudente comenzar la terapia antiagregante después de la intervención para minimizar el riesgo de hemorragia.
No hay diferencia de resultados entre los antiagregantes, aunque (dado el número de pacientes en los ensayos) es posible que haya pequeñas diferencias de eficacia no detectadas. En cualquier caso no hay criterios de eficacia para preferir un tratamiento sobre otro.
Sin embargo, estos datos son anteriores a la introducción de los bloqueantes de los receptores GP IIb/IIIa. Estos fármacos son aditivos al tratamiento antiagregante oral en angioplastia coronaria y mejoran los resultados en la prevención de episodios cardiovasculares.
Prevención de trombosis venosas profundas
El tratamiento antiplaquetario resulta en la disminución de unos 90 casos de trombosis y 17 casos de embolia pulmonar por 1000 tratamientos. Los resultados son parecidos para cirugía general y ortopédica y para pacientes no quirúrgicos de alto riesgo. En el lado negativo hay un aumento de 22/1000 tratamientos en episodios hemorrágicos leves a moderados y de 3/1000 tratamientos en casos que necesitaron transfusión. No parece haber aumento de episodios hemorrágicos mortales. Hay indicios, pero no demostración clara, de que la combinación AAS+dipiridamol es más efectiva que el AAS como preventivo de trombosis, pero la controversia real en estos momentos está en el papel de la prevención de la agregación plaquetaria respecto al tratamiento profiláctico con dosis bajas de heparina, que es procedimiento aceptado de prevención de trombosis venosas profundas. Los antiagregantes son más fáciles de usar, pero existe la impresión de que la acción protectora de la heparina es mayor y la experiencia con ella es más fiable. Hay estudios en marcha para confirmar los resultados y para determinar el posible beneficio adicional de la combinación de heparina y antiagregantes.
Prevención de la pre-eclampsia
La pre-eclampsia es asociada con una producción intravascular deficiente de prostaciclina, mientras que hay un aumento de la síntesis de tromboxanos. Esta observación ha conducido a algunos clínicos a sugerir el empleo de fármacos antiagregantes plaquetarios como terapia preventiva de la pree-eclampsia. De hecho, varios ensayos clínicos controlados han encontrado que el ácido acetilsalicílico es capaz de reducir en un 15% el riesgo de pre-eclampsia, con independencia del status de riesgo de la paciente o la edad de gestación. La mortalidad de los niños es reducida en un 14%, pero en cuanto al riesgo de parto prematuro, éste es reducido en apenas un 8% por los antiagregantes plaquetarios.
ENZIMAS
Los agentes fibrinolíticos o trombolíticos son sustancias que tienen por misión estimular el sistema fibrinolítico endógeno, de forma que sea éste el que disuelva el trombo formado. Un concepto importante es la tromboespecificidad, ya que existen agentes que estimulan
todo el sistema fibrinolítico del organismo (agentes tromboinespecíficos), por lo que destruyen la fibrina, pero también originan un estado de hiperfibrinolisis generalizada, lo cual es un factor predisponente para fenómenos hemorrágicos; mientras que otros agentes estimulan sólo la fibrinolisis allí donde existe fibrina, es decir, un trombo (agentes tromboespecíficos), respetando la fibrinolisis sistémica.
Cuanto más específico sea el fármaco, más trombolítico y menos fibrinolítico será. No obstante, en lenguaje coloquial, suelen utilizarse como sinónimos los términos de fibrinolítico y trombolítico.
Atendiendo a este concepto de especificidad, podemos clasificar a los agentes trombolíticos en tres grandes grupos, teniendo en cuenta también su mecanismo de acción:
A) Agentes inespecíficos o fibrinolíticos. Estimulan la fibrinolisis allí donde exista plasminógeno. Destacan la estreptoquinasa (SK) y la uroquinasa (UK).
– Estreptoquinasa: Polipéptido producido por el estreptococo beta-hemolítico grupo C. Se combina con el plasminógeno en cantidades equimoleculares y el complejo formado es capaz de activar la fracción restante de plasminógeno libre. Es el activador del plasminógeno más fácil de producir, y por tanto el más económico. Sus inconvenientes principales son:
- Es una proteína heteróloga, capaz de inducir la producción de anticuerpos. La presencia de anticuerpos (por exposición previa al medicamento o a una infección por estreptococos) implica la inactivación parcial, y por tanto cierta variabilidad en la respuesta.
- La necesidad de formar un complejo con el plasminógeno. Esto supone que una fracción significativa del plasminógeno plasmático no va a estar disponible para producir la lisis de la fibrina.
– Uroquinasa: Polipéptido de origen humano, obtenido a partir de cultivos de tejido renal embrionario. Activa directamente el plasminógeno (sin formar complejo con él) por ruptura del enlace entre los aminoácidos 560 y 561 de la cadena proteínica del plasminógeno. Es decir, estimula la hidrólisis de plasminógeno y lo convierte en plasmina, incluso sin participación de los activadores endógenos. La uroquinasa no es antigénica ni secuestra parte del fibrinógeno, pero es tres veces menos activa que la estreptoquinasa y por tanto precisa dosis proporcionalmente mayores, lo cual es un factor importante por las dificultades de producción del medicamento. Por este motivo es el agente trombolítico menos experimentado.
B) Agentes de especificidad intermedia. Se suele considerar como tal a la anistreplasa o APSAC. Es un complejo estreptoquinasa-plasminógeno, con el centro enzimático activo bloqueado por un radical ácido. Tras la administración, el radical ácido se desprende lentamente y queda libre el complejo capaz de activar el plasminógeno. Su relativa especificidad se debe a que la acilación del complejo requiere una deacilación orgánica, la cual se produce en el plasma; este proceso es lento, por lo que da más tiempo para estar en contacto con el trombo y menos para activar la fibrinolisis sistémica, pero siempre en términos muy relativos. Por otra parte, el plasminógeno de la anistreplasa es del tipo lys-plasminógeno, que se encuentra en mayor concentración en el ambiente circundante a un trombo, a diferencia del glu-plasminógeno, que es más frecuente en la circulación sistémica.
La anistreplasa tiene dos ventajas importantes sobre la estreptoquinasa. La primera es que lleva incorporado su propio plasminógeno para formar el complejo activador, y por tanto no necesita tomarlo del plasma. La segunda es que la semivida es muy larga comparado con el resto de los trombolíticos y no es necesario administrarla en infusión. Comparte con la estreptoquinasa los problemas inherentes a ser una proteína heteróloga.
C) Agentes específicos o trombolíticos. Son aquellos que activan principalmente la fibrinolisis allí donde hay fibrina, es decir, en un trombo y no en la circulación sistémica. Destacan el activador tisular del plasminógeno recombinante (rtPA o alteplasa) y sus variantes, junto con la prouroquinasa (proUK).
– Formas recombinantes de Activador tisular de plasminógeno (tPA): Actúan de forma similar a como lo hace el tPA endógeno, es decir, necesita de forma imprescindible su unión a la fibrina para acoplarse al plasminógeno y transformarlo en plasmina, es decir, sólo se activaría la fibrinolisis si existe fibrina.
- Alteplasa: Es el factor tisular de activación del plasminógeno (tPA) obtenido por ingeniería genética. La alteplasa tiene una afinidad relativamente débil hacia el plasminógeno circulante, pero la afinidad se ve muy potenciada en presencia de fibrina. El efecto activador del plasminógeno se produce fundamentalmente en el punto de formación del trombo. La alteplasa se desarrolló buscando un medicamento que no produjese la lisis generalizada del plasminógeno con el consiguiente cuadro de hipocoagulabilidad. Sin embargo la semivida es muy corta y generalmente se usa junto con heparina para prevenir la reoclusión. Esto contrarresta su ventaja principal. Por el mismo motivo el tiempo de infusión es relativamente largo.
- Reteplasa: Es un mutante no glucosilado del Activador Tisular del Plasminógeno (alteplasa). Las modificaciones estructurales presentes parecen provocar un menor grado de unión a la fibrina, una mayor potencia trombolítica y una semivida más prolongada. Algunos estudios realizados sobre animales de experimentación parecen sugerir que la reteplasa es mejor que otros trombolíticos anteriores (alteplasa, anistreplasa, estreptokinasa). También en estudios clínicos parece haberse confirmado un mejor papel terapéutico, con mayores porcentajes de pacientes con patencia arterial coronaria total y reperfusión completa que la alteplasa, con la correspondiente reducción de la necesidad de intervenciones coronarias adicionales. Por el contrario, se han obtenido similares resultados con estreptokinasa.
- Tenecteplasa. Se trata de una glucoproteína recombinante que consiste en una forma modificada de tPA natural. Concretamente, se han modificado tres sitios de la molécula natural mediante mutegénesis. La tenecteplasa presenta unos niveles de eficacia muy similares a los de alteplasa. Tan sólo se aprecia una leve reducción, aunque estadísticamente significativa, de las complicaciones hemorrágicas no cerebrales y, consecuentemente, una menor necesidad de transfusiones sanguíneas.
― proUK. Es una molécula inactiva, que necesita transformarse en uroquinasa (UK), pero este paso tiene como cofactor fundamental a la fibrina, de ahí que se produzca casi selectivamente alrededor de un trombo.
TABLA III. BENEFICIOS CLÍNICOS DE LA TERAPIA TROMBOLÍTICA
| INDICACIÓN | % DE REPERFUSIÓN | PERIODO ÓPTIMO TRAS OCLUSIÓN |
| Embolia pulmonar | 80-90 | menor de 48 horas |
| Trombosis venosas profundas | 60 | menor de 5 días |
| Oclusión arterial periférica | 40 (sistémico)
60 (regional) |
menor de 14 horas |
| Infarto de miocardio | 50-70 (sistémico)
75 (intracoronario) |
menor de 2 horas |
TERAPIA TROMBOLÍTICA EN INFARTO DE MIOCARDIO
Numerosos ensayos han demostrado que la terapia trombolítica puede aumentar significativamente la supervivencia si se administra antes de que se produzca la necrosis de la zona privada de irrigación.
La aplicación IV de agentes trombolíticos tiene el inconveniente de producir la activación global del plasminógeno y por tanto un defecto generalizado de la coagulación. Los nuevos productos son fruto de intentos de conseguir acción lítica limitada al punto de trombosis. Esta propiedad parece tener poco interés desde el punto de vista práctico. El peligro de hemorragias no disminuye porque realmente no hay especifidad hacia el trombo: la acción se ejerce también sobre la fibrina de los tapones hemostáticos.
Por otro lado, el estado de hipocoagulabilidad no es tan indeseable como se pensó en un principio. El principal problema de la fibrinolisis es evitar la reoclusión del vaso (de las arterias coronarias, en el infarto). Para ello, estos fármacos tienen que ser administrados con anticoagulantes y, cada vez más, antiagregantes plaquetarios, para minimizar este fenómeno, con lo cual se pierde cualquier ventaja relacionada con el efecto general sobre la coagulación.
La eficacia del tratamiento depende del tiempo entre el inicio de la sintomatología y la administración del trombolítico. Existe cierto efecto protector hasta las 12 horas del comienzo del infarto, aunque la mayor parte de los tratamientos están indicados hasta las primeras seis horas, y es posible (pero no está probado) que se obtenga cierto beneficio en tratamientos entre las 12 y las 24 horas.
En general, el aumento de supervivencia se produce en cuadros caracterizados por electrocardiogramas (ECG) con segmento ST elevado o bloqueo fascicular sin depresión ST elevado o bloqueo fascicular sin depresión ST. No hay diferencias en pacientes con otras anormalidades del ECG o con ECG normal, aunque en muchos casos falta de significación estadística pudiera deberse a la menor mortalidad general. La eficacia terapéutica de todos los trombolíticos es parecida, aunque los productos más modernos parece presentar menores riesgos hemorrágicos graves.
LOS TROMBOLÍTICOS EN URGENCIAS EXTRAHOSPITALARIAS
Los trombolíticos tienen en España (no en otros países) la clasificación de «medicamento de uso hospitalario». Sin embargo la tabla IV muestra claramente que la efectividad aumenta en función de la rapidez de la aplicación. De aquí el interés de la administración extrahospitalaria.
Se han hecho ensayos en uso extrahospitalario (unidades móviles de urgencias coronarias y similares) que muestran que la práctica es bastante segura si la ambulancia está dotada de equipo de diagnóstico y para el tratamiento de arritmias, y de personal bien adiestrado en su uso. Pero las estadísticas de supervivencia no mejoran mucho.
La causa fundamental es que el tiempo promedio desde inicio de síntomas hasta la atención al paciente no baja de las 1,5-2 horas, y la diferencia con el ingreso hospitalario es del orden de 45 minutos a una hora. En estas circunstancias es lógico que las diferencias de resultados sean pequeñas.
Un motivo importante del retraso parece ser el tiempo que tarda el paciente en decidirse a solicitar asistencia médica. Las campañas de concienciación pública tienen un efecto transitorio y afectan más al número de falsas alarmas que a la rapidez de decisión en los casos reales, donde influye un factor de auto-denegación de la gravedad del cuadro. La opinión actual es que en el tratamiento del infarto agudo no importa tanto dónde como cuándo. El problema es de planificación sanitaria. Deben establecerse los medios apropiados («vías rápidas» en procedimientos de ingreso hospitalario, servicios extrahospitalarios donde sea necesario) para que el tiempo entre la llamada al sistema sanitario y la administración del trombolítico no supere un valor prefijado (por ejemplo 90 minutos).
OTRAS APLICACIONES DE LOS TROMBOLÍTICOS
Trombosis cerebral: La teoría es la misma que en el infarto de miocardio, es decir que la rápida disolución del trombo y la reapertura del vaso afectado minimiza la necrosis tisular, aumenta la probabilidad de supervivencia y disminuye la gravedad de las secuelas.
Existen sin embargo dos problemas que no se dan en la trombosis coronaria:
- Durante el tiempo de oportunidad del tratamiento (arbitrariamente establecido en 6 horas desde la oclusión) hay que hacer un diagnóstico diferencial entre accidente vascular trombótico y hemorrágico, lo cual requiere el empleo de tomografía computerizada.
- Las hemorragias inducidas por los trombolíticos son una complicación más frecuente y más grave en la terapia cerebral que en la coronaria.
A pesar de ello, la aplicación en trombosis cerebral es la indicación más estudiada de los fibrinolíticos después de infarto de miocardio. Se han publicado recientemente los resultados de varios ensayos extensos, pero la mayoría acabaron de forma distinta a la planificada y es difícil sacar en estos momentos una conclusión definitiva. Las conclusiones provisionales son: 1.- La terapia trombolítica produce un aumento significativo de la mortalidad en el primer mes (razón de la suspensión de varios ensayos) pero mejoría significativa entre tres y seis meses, tanto de mortalidad como de secuelas neurológicas. 2.- Es posible que haya una diferencia importante en el resultado entre los pacientes tratados dentro de las primeras tres horas desde la trombosis, respecto a los tratados entre tres y seis horas.
El dilema terapéutico de si es aceptable la mayor mortalidad inicial a cambio del mejor pronóstico de los supervivientes no se puede resolver con los datos actuales y tendrá que esperar posteriores estudios. Es posible que la solución radique en precisar mejor las condiciones de tratamiento y refinar los criterios de selección de pacientes.
Trombosis arterial periférica: En comparación con las técnicas quirúrgicas, la terapia trombolítica no parece mejorar los resultados clínicos, precisando incluso más tiempo de aplicación (una media de 24 h) y produce más complicaciones hemorrágicas. Por todo ello, no puede contemplarse a la terapia trombolítica como tratamiento de primera línea en la trombosis arterial aguda periférica (Porter JM. Thrombolysis for acute arterial occlusion of the legs. N Engl J Med 1998; 338; 1148-9).
Trombosis venosas profundas: No se suelen usar salvo en casos muy seleccionados. Por lo general los trombos son voluminosos y requieren demasiado tiempo de actuación del trombolítico, con considerable riesgo de hemorragia. Los ensayos clínicos controlados no han mostrado hasta el momento beneficio en términos de mortalidad o secuelas crónicas en las extremidades.
Embolia pulmonar: Es una práctica clínica habitual administrar trombolíticos en cuadros severos de embolia pulmonar, pero no hay estudios bien controlados sobre la repercusión del tratamiento en la mortalidad.
OTROS ANTITROMBÓTICOS
HIRUDUINAS
Los derivados de origen recombinante de la hirudina, el anticoagulante producido por las sanguijuelas (Hirudo medicinalis) producen un potente efecto anticoagulante, consecuencia del bloqueo de la actividad trombogénica de la trombina, mediante la formación de un complejo equimolecular con esta última, de carácter no covalente. Esto conduce a una inhibición directa de todas las acciones de la trombina, tanto de la libre como de la ligada a los coágulos, lo cual la diferencia de la heparina.
Las hirudinas recombinantes no requieren para su actuación la participación de factores endógenos y actúa de forma independiente de la antitrombina III y del cofactor II de la heparina. Todo ello se traduce en un efecto sobre la coagulación más estable que el conseguido con heparina. Por el momento, se han registrado en España desirudina y lepirudina, ambas derivadas de la 63-desulfohirudina, aunque la última presenta los dos primeros aminoácidos cambiados con respecto a la hirudina natural.
Desirudina ha sido autorizada en España para la prevención de trombosis venosa profunda en pacientes sometidos a cirugía electiva de prótesis de rodilla y de cadera. En esta indicación, desirudina parece producir resultados profilácticos notablemente mejores que la heparina convencional y las heparinas de bajo peso molecular, al reducir el riesgo de trombosis venosa profunda en un 50-70% en relación a la heparina no fraccionada, y en un 30% frente a enoxaparina. La diferencia es aun más marcada en la prevención de las trombosis proximales (40-90%).
Lepirudina ha sido autorizada para su uso en pacientes con trombocitopenia inducida por heparina. Aunque la aparición de trombocitopenia no es infrecuente durante los primeros días de tratamiento con heparina, en la mayoría de los casos se trata de fenómenos de carácter leve y transitorio; sin embargo, el 1-2% de los pacientes tratados con heparina durante más de cuatro días desarrollan un cuadro trombocitopénico mucho más grave, con una tasa de mortalidad relativamente alta. El fármaco ha demostrado un elevado nivel de eficacia en esta indicación, produciendo una rápida recuperación del recuento plaquetario en más del 90% de los pacientes, y manteniendo una anticoagulación eficiente en torno al 80% de ellos. Las consecuencias de estas acciones son una fuerte reducción de la mortalidad en los pacientes, que puede cifrarse en el 50% al cabo de un mes de tratamiento, así como de la incidencia de complicaciones tromboembólicas, con una reducción superior al 70% en una semana.
OTROS
La proteína C humana es una glucoproteína de cadena doble y 62 kD de peso molecular, sintetizada por el hígado y dependiente de la vitamina K. Normalmente, circula en la sangre como un zimógeno inactivo. Es activada por el complejo trombina/trombomodulina (la trombomodulina es una proteína receptora de membrana presente en las células endoteliales). La activación de la proteína C conduce al desarrollo de una actividad de tipo serinproteasa, que determina una acción sobre los factores Va y VIIIa, a los que destruye, actuando la proteína S como cofactor. Su semivida plasmática es muy corta (7-9 horas).
La vía metabólica de la proteína C supone un mecanismo natural de control del sistema de coagulación sanguíneo, que impide una excesiva coagulabilidad. Por consiguiente, la deficiencia, congénita o adquirida, de proteína C da lugar a un incremento de la coagulación que determina la generación de trombina y, en última instancia, la formación de coágulos intravasculares (trombosis).
La proteína C humana se utiliza en el tratamiento de la púrpura fulminante y de la necrosis de piel inducida por cumarinas en pacientes con deficiencia congénita grave de proteína C, así como en profilaxis a corto plazo en pacientes con deficiencia congénita grave de proteína c si es inminente la cirugía o terapia invasiva, mientras se inicia la terapia cumarínica o cuando ésta no es suficiente o no es posible .
TRATAMIENTO FARMACOLÓGICO DEL INFARTO DE MIOCARDIO
INTERVENCIÓN EN INFARTO AGUDO
La elevada mortalidad en las primeras horas y el conocimiento de que la intervención que logre limitar el tamaño del infarto va a tener una gran influencia en la supervivencia han despertado interés hacia los tratamientos de urgencia y preventivos en el infarto agudo.
Sin embargo sigue habiendo obstáculos muy importantes a estas técnicas como son: la dificultad de un diagnóstico preciso, la casi inevitabilidad de un periodo de varias horas hasta que el enfermo recibe asistencia sanitaria y los riesgos inherentes a muchos medicamentos usados en cuadros agudos.
La mayoría de los tratamientos que vamos a relacionar tienen escasa eficacia si se aplican después de 6-8 horas de comenzado el infarto. No se incluyen en esta guía técnicas no farmacológicas, como la cardioversión eléctrica o la angioplastia, cuyo interés en el tratamiento del infarto no puede ser minimizado.
TRATAMIENTO DE URGENCIA PREVIA A HOSPITALIZACIÓN
INTERVENCIÓN |
|
| TRATAR EL DOLOR
Morfina Meperidina |
La morfina por vía IV es el tratamiento de elección, no sólo por su acción analgésica sino por sus efectos hemodinámicos. Administrar 3-5 mg cada 5-10 minutos hasta obtener el efecto analgésico deseado sin excesiva sedación o hipotensión. La meperidina (25-75 mg, IV) puede ser preferible en presencia de bradicardia, náuseas o vómitos, o en antecedentes de litiasis biliar o colecistitis. Algunos recomiendan administrar un antiemético IV (ej. metoclopramida) junto con el analgésico. |
| ÁCIDO ACETILSALICLICO | Los ensayos con antiagregantes plaquetarios han demostrado que el AAS reduce significativamente la mortalidad y la incidencia de episodios cardiovasculares no mortales y el beneficio es aditivo a la terapia trombolítica. Respetando las contraindicaciones clásicas del AAS, ésta es la terapia preventiva con menos riesgo de complicaciones que puede darse fuera del hospital, y se ha convertido en un elemento básico de los protocolos de tratamiento. El paciente puede masticar un comprimido de 500 mg de AAS para obtener rápidamente niveles plasmáticos adecuados. El seguimiento se hace a dosis menores (150 a 300 mg). |
| CORREGIR o PREVENIR ARRITMIAS | La bradicardia (menos de 50 latidos/minuto) especialmente si va acompañada de hipotensión, puede responder a 0,6 mg IV de atropina repetida si es necesario hasta un máximo de 2 mg.
Puede ser también un tratamiento de último recurso en asístoles refractarias. Para arritmias ventriculares puede administrarse lidocaína en bolo IV de 50 mg, repetido si es necesario a intervalos de 5 minutos hasta un máximo de 225 mg. La administración profiláctica rutinaria de 400 mg de lidocaína IM es una práctica controvertida y poco usada. Los enayos han demostrado que la incidencia de fibrilación ventricular y asístole baja significativamente del 1,4% al 0,2%, pero en cambio la mortalidad general registró un alza no significativa del 11%. Las dificultades de diagnóstico hacen que un porcentaje de pacientes con falsa sintomatología de infarto reciban el medicamento. |
| BETABLOQUEANTES
(Ver grupo C07A) |
Ensayos muy extensos han demostrado que se obtienen aumentos significativos de supervivencia (15% o más en 1-2 semanas) con los regímenes terapéuticos siguientes:
– Metoprolol 15 mg IV en las primeras 12 horas seguido de 100 mg 2 veces al día por vía oral. – Atenolol 5-10 mg IV en las primeras 5 horas seguido de 100 mg al día por vía oral. La importancia de la administración temprana aboga por la intervención en urgencia pre-hospitalaria, pero por otro lado ésta no es muy conveniente porque los betabloqueantes están contraindicados en cuadros que se asocian con bastante frecuencia al infarto: insuficiencia cardíaca, bloqueo AV, hipotensión, bradicardia. Los datos clínicos proceden todos de tratamientos hospitalarios, las incertidumbres de las urgencias prehospitalarias no han sido valoradas adecuadamente. En España nos encontramos con la dificultad adicional de que los preparados inyectables de betabloqueantes tienen la clasificación legal de «uso hospitalario». En cualquier caso un tratamiento de urgencia por personal experimentado podría ser beneficioso en cuadros caracterizados por hipertensión y taquicardia. |
| CORREGIR LA INSUFICIENCIA VENTRICULAR IZQUIERDA AGUDA | Puede tratarse con furosemida IV (20-40 mg) y administrando oxígeno (ver más abajo). |
| ADMINISTRAR OXÍGENO | Por cánula nasal o mascarilla. 3-4 litros/minuto. Suele usarse rutinariamente, pero no está establecida la utilidad en casos de infarto no complicado. |
TRATAMIENTO EN HOSPITAL
INTERVENCIÓN |
|
| TERAPIA TROMBOLÍTICA |
Ver comentario general en la introducción al GRUPO B01AD más arriba.
La trombolisis, junto con AAS, es la base del tratamiento de urgencia del infarto. La práctica más usual es administrar inmediatamente un trombolítico a los pacientes que presenten sintomatología de infarto y elevación del segmento ST del ECG, si no hay contraindicaciones formales. La edad avanzada no constituye contraindicación por sí sola. |
| BETABLOQUEANTES | Ver comentario en Tratamiento Pre-Hospitalización arriba.
Como ocurre con otros tratamientos limitantes del tamaño del infarto, el uso hospitalario es un mal compromiso entre la necesidad de controlar las condiciones de aplicación y el interés de iniciar la terapia lo antes posible. Los beneficios que se obtienen son del 28% de disminución de mortalidad (principalmente en las primeras 48 horas), 18% de disminución del reinfarto y 15% de reducción de parada cardíaca. El éxito de los trombolíticos ha desplazado a los betabloqueantes de los protocolos de tratamiento del infarto agudo, pero los tratamientos son compatibles, aunque no está claro hasta qué punto los beneficios son aditivos. En cualquier caso se recomienda hoy día el tratamiento con betabloqueantes (IV seguido de administración oral) a todos los pacientes donde no estén específicamente contraindicados (menos de 60 latidos/minuto, presión sistólica<100 mmHg, insuficiencia ventricular izquierda moderada a severa, bloqueo severo de conducción AV1, asma o enfermedad respiratoria obstructiva). Los criterios de exclusión pueden llegar a afectar a la mitad de los casos. |
| MAGNESIO IV | La utilidad del magnesio IV (8 mmol en bolo IV durante 15 minutos, seguida de infusión IV de 65 mmoles en 24 horas) está en discusión.
Varios ensayos en pequeña escala y uno extenso y bien controlado (LIMIT-2) han mostrado disminución estadísticamente significativa de la mortalidad a las 4 semanas y a los 2,7 años (21% de mortalidad cardiovascular, 16% de mortalidad por todas las causas). Sin embargo el muy extenso ensayo ISIS-4 no ha encontrado beneficio en términos de mortalidad. Hay diferencias en los ensayos que pueden ser relevantes. En el estudio positivo LIMIT-2 el magnesio se usó antes que los trombolíticos (mediana: 3 horas desde el comienzo de síntomas), mientras que en ISIS-4 se usó después de haber conseguido la reperfusión con el trombolítico (mediana: 8 horas desde los síntomas). Puesto que los trombolíticos se han convertido en el tratamiento estándar del infarto, el papel del magnesio debe definirse con relación a ellos. Por los datos actuales, es poco útil después de un tratamiento trombolítico. La hipótesis de que es interesante antes del trombolítico necesita confirmación. En cualquier caso, la experimentación animal indica que el tiempo desde la hipoxia es crítico en la eficacia. Posiblemente le mejor opción es tenerlo en cuenta en casos donde la trombolisis esté contraindicada o cuando se presenten cuadros de arritmia e hipomagnesemia. |
| CORREGIR/PREVENIR ARRITMIAS | La atropina y la lidocaína son los dos puntales del tratamiento antiarrítmico, pero pueden usarse otros medicamentos en caso necesario. Ver grupo C01B ANTIARRÍTMICOS.
Con monitorización cardíaca y administrada por infusión intravenosa, el uso de lidocaína es menos problemático que en las urgencias extrahospitalarias. La profilaxis de fibrilación ventricular no se recomienda ya. Los ensayos clínicos muestran que la reducción en la incidencia de fibrilación es contrarrestada por aumento de incidencia de asístole de tal forma que la mortalidad no experimenta gran variación. |
| VASODILATADORES | La nitroglicerina sublingual o IV tiene interés para disminuir el trabajo cardíaco por disminución de la resistencia vascular. Es útil sobre todo cuando hay insuficiencia congestiva, pero se ha comprobado disminución de mortalidad en todos los casos. Debe evitarse su uso en caso de hipotensión o bradicardia.
Algunos clínicos administran nitroglicerina sublingual a todos los pacientes con presión sistólica superior a 90 mmHg. La administración IV debe considerarse especialmente para pacientes con dolor precordial recurrente o infartos relativamente grandes. Se hace lentamente (para evitar bradicardia e hipotensión) y vigilando cuidadosamente la respuesta hemodinámica y clínica. Una pauta puede ser: comenzar con una infusión de 10-20 mcg/min, que puede aumentarse en 5-10 mcg/min cada 5-10 minutos hasta alcanzar la respuesta adecuada. La dosis de mantenimiento suele situarse entre 50 y 300 mcg/min. El nitroprusiato sódico tiene menos aceptación por el riesgo de producir un «fenómeno de robo» en detrimento de la irrigación de la zona infartada. Los ANTAGONISTAS DEL CALCIO han dado peores resultados en aplicación clínica que en investigación animal y tienen poco interés en el tratamiento del infarto. El diltiazem es el único medicamento del grupo con alguna utilización. |
| ANTICOAGULANTES | No existe gran evidencia que apoye la utilidad de la anticoagulación en el tratamiento del infarto, pero es una práctica rutinaria en el tratamiento hospitalario que se ha revalorizado con la extensión de la terapia trombolítica, uno de cuyos principales problemas es evitar la posterior reoclusión del vaso.
Las dosis bajas de heparina (5000 UI por vía SC al día mientras el paciente esté en cama) puede prevenir la trombosis venosa o la embolia pulmonar en pacientes de riesgo moderado. La anticoagulación completa tiene interés en pacientes de alto riesgo de trombosis venosa o de desarrollar trombos en la zona infartada. Se usa también como complemento de la terapia trombolítica, sobre todo con alteplasa. |
| TRATAR LA INSUFICIENCIA CARDIACA CONGESTIVA |
La primera línea de tratamiento es oxígeno y furosemida IV (ver arriba). Procurar limitar la dosis y duración del diurético para evitar la hipovolemia. La nitroglicerina IV es el siguiente escalón. Para cuadros refractarios al tratamiento anterior suelen usarse agentes inotropos como dopamina y dobutamina (ver GRUPO C01A2A) por vía IV. Las propiedades hemodinámicas difieren algo: la dopamina se utiliza preferentemente cuando existe hipotensión que se beneficie de la vasoconstricción que el fármaco produce. La dobutamina es útil sobre todo si hay congestión pulmonar.
La digoxina tiene en estos cuadros un papel limitado. Los inhibidores de la angiotensina-convertasa pueden aumentar la supervivencia de pacientes con disfunción ventricular izquierda (ver el apartado tratamiento postinfarto). |
| TRATAR LA FIEBRE | Paracetamol o AAS cada 4-6 horas durante 48-72 horas. |
| TRATAR LA ANSIEDAD |
Benzodiazepinas (ver GRUPO N05B). Por ejemplo: 2,5 a 5 mg de diazepam (o 2-3 mg de lorazepam) dos a tres veces al día. |
| EVITAR ESFUERZOS EN LA DEFECACIÓN | Usar docusato sódico u otro laxante suave (ver GRUPO A06). |
TABLA V. BENEFICIO ESTIMADO DE DIFERENTES INTERVENCIONES FARMACOLÓGICAS EN INFARTO AGUDO
| INTERVENCIÓN | Disminución de mortalidad por 1000 tratamientos |
| Trombolítico antes de 6 horas.
Ácido acetilsalicílico vía oral. Inhibidor de angiotensina-convertasa oral. Beta-bloqueante IV seguido de oral. |
20-35
20-25 5-8 6 |
TABLA VI. INTERVENCIÓN FARMACOLÓGICA POSTINFARTO
| MEDICAMENTO
Beneficio por 1000 tratamientos |
COMENTARIO |
|
| PREVENCIÓN DEL REINFARTO | Antiagregantes plaquetarios
– 40 eventos cardiovasculares * |
La acción de los antiagregantes en el postinfarto se trata con detalle en la introducción del grupo B01.
El ácido acetilsalicílico es el antiagregante de elección y uno de los puntales del tratamiento postinfarto. |
| Anticoagulantes | El efecto protector de los anticoagulantes es del mismo orden o inferior al de los antiagregantes plaquetarios. Puesto que el riesgo de episodios hemorrágicos es muy superior no hay justificación para usar anticoagulantes, salvo riesgo significativo de trombosis (por ejemplo, infartos extensos anteriores trasmurales). | |
| PREVENCIÓN DE MUERTE POR ARRITMIA | Betabloqueantes
– 15 muertes – 5 infartos no mortales |
El mecanismo de acción es fundamentalmente antiarrítmico y por disminución de la demanda de oxígeno, pero también hay un factor de disminución del reinfarto por razones poco conocidas. Hay demostración estadísticamente significativa de eficacia para el propranolol, metoprolol y timolol pero es probable que sea una propiedad general del grupo.Los mejores resultados parecen darse cuando quedan arritmias o disminución leve de la función ventricular (están contraindicados en disfunción moderada o grave) pero se ven beneficios en cuadros de cualquier tipo y la opinión más generalizada es que deben administrarse en todos los casos donde no estén contraindicados.
Las dosis usuales son 100 mg de metoprolol o 10 mg de timolol (ambos 2 veces al día), o 180-240 g de propanolol en 2-3 tomas. El tratamiento debe continuarse indefinidamente, pero algunos clínicos tienen la impresión de que la diferencia de supervivencia a los doce meses es baja, y por tanto el tratamiento puede suspenderse al año si el paciente no lo tolera bien. |
| Antiarrítmicos | El uso de antiarrítmicos se basa en la apreciación de que las contracciones ventriculares prematuras (CVP) constituyen un indicativo de riesgo elevado de arritmias ventriculares graves o muerte súbita postinfarto. Sin embargo los ensayos clínicos hechos con antiarrítmicos de la clase I (que habían demostrado experimentalmente capacidad para suprimir las CVP) mostraron mayor mortalidad en el grupo tratado que en el control. Lo mismo ha ocurrido con el antiarrítmico de clase III Sotalol. El fenómeno se debe probablemente a propiedades proarrítmicas de los medicamentos. El único que permanece en estudio es la amiodarona. No parece aconsejable usar antiarrítmicos en la prevención de la mortalidad post-infarto. | |
| PREVENCIÓN DE
INSUFICIENCIA CARDIACA |
Inhibidores de la angiotensina-convertasa – 5 a 40 muertes – 9 infartos – 16 casos de insuficiencia cardíaca congestiva |
Se han hecho extensos ensayos con captoprilo, enalaprilo, ramiprilo y lisinoprilo, pero las conclusiones son extensibles muy probablemente a todos los IECA.
Los IECA son especialmente beneficiosos en pacientes con disfunción ventricular izquierda (se previenen entre 20 y 45 muertes por 1000 tratamientos). La administración sistemática a pacientes no hipotensos dentro de las 24 horas del infarto es también efectivo pero mucho menos (5 a 8 muertes por 1000 pacientes, ver tabla V) y existe controversia sobre si tiene entidad suficiente para justificar el tratamiento rutinario sin verificar si hay insuficiencia ventricular. |
* Evento cardiovascular es: muerte de causa cardiovascular, reinfarto no mortal o accidente vascular cerebral no mortal.
ESTRATEGIAS DE TRATAMIENTO POSTINFARTO
Tres grupos de medicamentos han demostrado, en ensayos extensos bien controlados, que aumentan la supervivencia postinfarto: los antiagregantes plaquetarios, los betabloqueantes y los inhibidores de la angiotensina convertasa (IECA).
Pero las condiciones de los ensayos han sido muy variadas y aunque los meta-análisis permiten establecer -con fiabilidad relativa- estimaciones de beneficio terapéutico que pueden servir para comparar tratamientos a nivel individual, no está claro hasta qué punto estos beneficios son aditivos. No existen estrategias definidas para terapias combinadas.
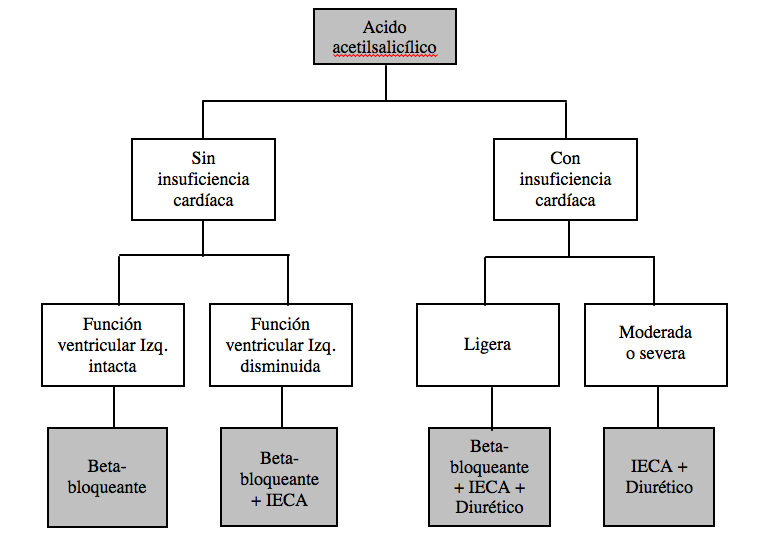
En estos momentos, el tratamiento básico postinfarto es el ácido acetilsalicílico. A partir de aquí puede seguirse una estrategia conservadora consistente en adicionar un segundo medicamento dependiendo de la naturaleza de las secuelas (beta-bloqueantes si predominan cuadros arrítmicos, IECA si disfunción ventricular o clara insuficiencia).
Otros clínicos preconizan una estrategia más agresiva, maximizando el tratamiento farmacológico como se indica, por ejemplo, en el esquema siguiente:
El contenido aquí mostrado corresponde a BOT (base de datos de medicamentos de España 2002), que aunque está relacionado con medicamentos de uso humano, puede resultar muy útil para la medicina veterinaria de pequeños animales.